BLOG
Introducing OrbMol-v2: A Neural Network That Outperforms the Most-Cited Method in Chemistry
June 2, 2026 · Orbital

We're excited to announce OrbMol-v2: a new universal molecular machine learning interatomic potential (MLIP) that beats hybrid GGA functionals like PBE0 and B3LYP at computing molecular energies and forces at 6 orders of magnitude lower cost. These functionals underpin much of modern computational chemistry: the foundational B3LYP papers alone have been cited around 100,000 times each.
The DFT crossover
We evaluate our new model using GSCDB, the most diverse and rigorous open benchmark for molecular DFT (Liang & Head-Gordon, J. Chem. Theory Comput. 21, 12601, 2025). GSCDB measures errors as a normalized error ratio (NER): the model's error divided by a reference error from the best hybrid DFT functionals, so NER = 1 means matching the best DFT, and NER < 1 means beating it.
To compare DFT functionals and MLIPs fairly, we evaluate every method on a subset of GSCDB that excludes single atoms. On this common basis:
- OrbMol-v2 achieves NER 1.62, 21% more accurate than the next best MLIP.
- It has only one reaction with absolute error above 100 kcal/mol across the entire benchmark (OrbMol-v1 baseline: 59; UMA-s-1.2 and MACE-POLAR-M: 6 each). With this near-absence of outliers, MLIPs are becoming viable as a first-line replacement for DFT.
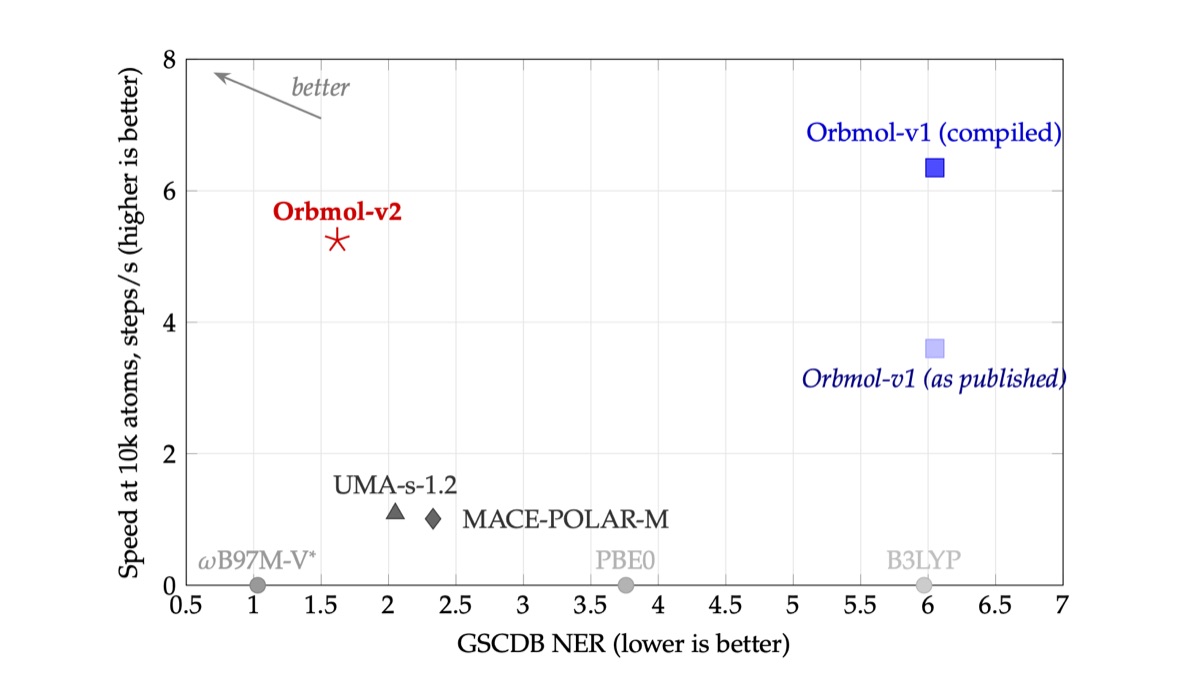
Figure 1: Accuracy-speed tradeoff for universal MLIPs and DFT functionals on GSCDB. Grey points show DFT functional NER values on the same filtered basis, making the DFT and MLIP comparison apples to apples. OrbMol-v2 achieves NER 1.62, outperforming widely used hybrid GGA functionals while remaining ~5x faster than competing MLIPs at 10,000 atoms on a single H100 GPU. OrbMol-v2 is faster than OrbMol-v1 as published due to improved compilation (which also accelerates OrbMol-v1) and more than offsets the overhead from the long-range electrostatics calculation.
Why it's so fast
The most exciting aspect of this model is that the long-range interactions cost very little compared with the backbone. Other approaches to MLIP electrostatics have used iterative charge-equilibration schemes that slow inference down. In OrbMol-v2, the PME-based Coulomb term runs at similar speed to the backbone alone. When combined with improvements in model compilation, OrbMol-v2 is substantially faster than OrbMol-v1 and 5x faster than comparable models on a 10,000-atom system. This is made possible by NVIDIA ALCHEMI Toolkit-ops, which provides fast GPU-accelerated batched PME for computing long-range electrostatic interactions, together with recent NVIDIA ALCHEMI Toolkit-ops neighbour-list speedups that enable fast batched inference.
What's new
Training data is crucial too. OrbMol-v2 is trained on a large molecular training corpus: OMol-25 and OPoly, both at the ωB97M-V/def2-TZVPD level of theory. OMol-25 contains 100M+ DFT calculations spanning small molecules, biomolecules, metal complexes, ions, electrolytes, variable charge states, and spin multiplicities; OPoly adds polymer fragments and organic-crystal geometries.
We add three components on top of the OrbMol-v1 backbone:
- Per-atom predicted latent charges constrained to sum to each system's total charge.
- An explicit Coulomb energy term driven by these charges (direct sum for molecules; Particle Mesh Ewald for periodic systems), giving the model long-range electrostatics.
- A charge-conditioned energy head that conditions each atom's short-range energy contribution on the predicted latent charge features.
Together, these give OrbMol-v2 a physically motivated treatment of electrostatics at minimal computational overhead.
Ions and ionic dissociation
A direct test of long-range interactions is the NaCl dimer dissociation curve. Models without explicit long-range interactions produce a shallow potential that decays to zero beyond their interaction cutoff: qualitatively wrong wherever ions can separate. OrbMol-v2 produces a deep ionic binding well with the correct long-range -1/r tail. It also predicts the correct physical charges at long range as well as the physically expected charge transfer as they come together. It does so without relying on explicit charge labels.
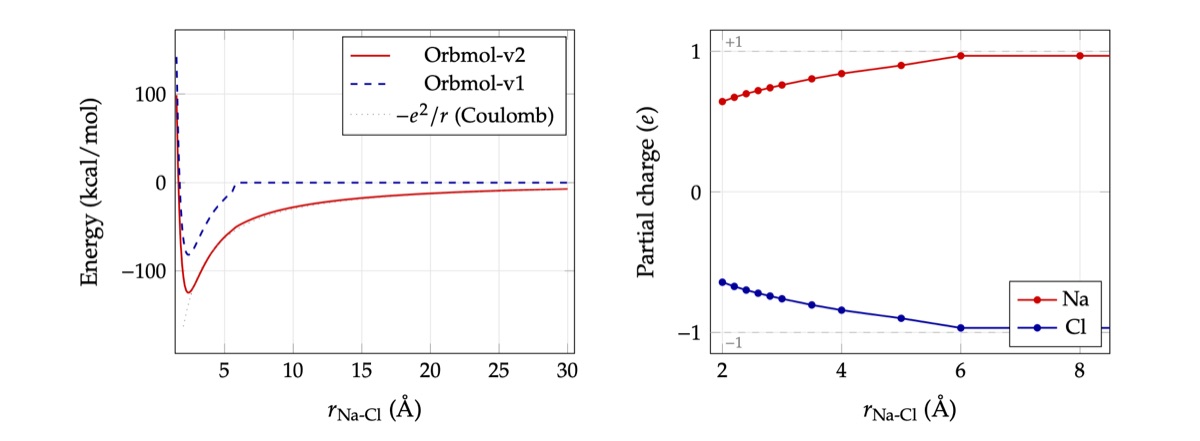
Figure 2: LHS: NaCl dimer binding energy. OrbMol-v2's explicit Coulomb term captures the ionic bond with the correct long-range -1/r tail; OrbMol-v1 (no electrostatics) goes to zero beyond its GNN cutoff. Dotted line: exact Coulomb potential for +/-1 e charges. RHS: charge transfer as the two ions come together showing correct learned charges at long range with charge transfer as they come together as physically expected.
Built with agents
The prototype implementation of the electrostatics module, benchmarks, simulations, and documentation was initially performed by LLM agents iterating on top of the existing Orb code base. Final production required extensive expert human review and improvement before release, and a mature, well-developed codebase to iterate on top of was critical. Nonetheless, this result demonstrates the potential for increasingly automated computational chemistry research, with much faster iteration and testing of new ideas.
The combination of fast, accurate MLIPs and capable LLM agents is central to the future of computational chemistry, with agents reasoning about, designing, and orchestrating model training and simulation campaigns with increasing autonomy.
Try it
The model and code are released as part of the open-source Orb family at orb-models.
Additional metrics
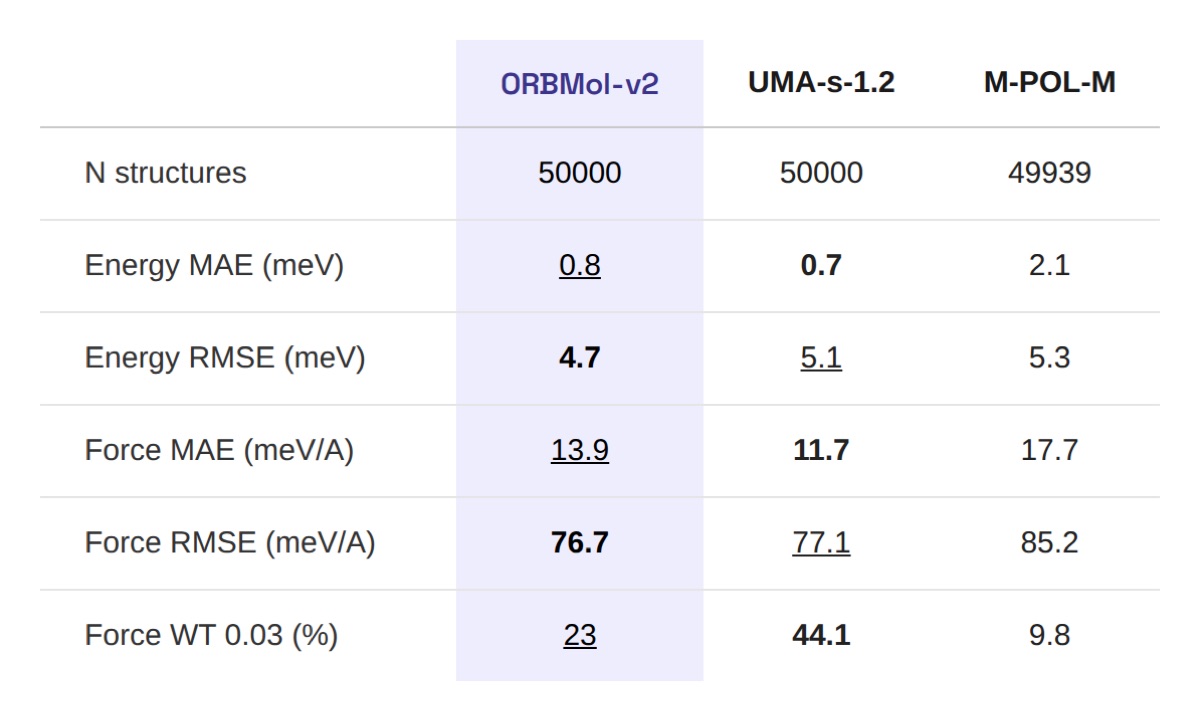
Table 1: Validation set results on 50,000 structures sampled from the OMol-25 validation set. Energy errors are per atom. Force WT is the fraction of structures with all force components within 0.03 eV/Å. Bold = best, underline = second best.
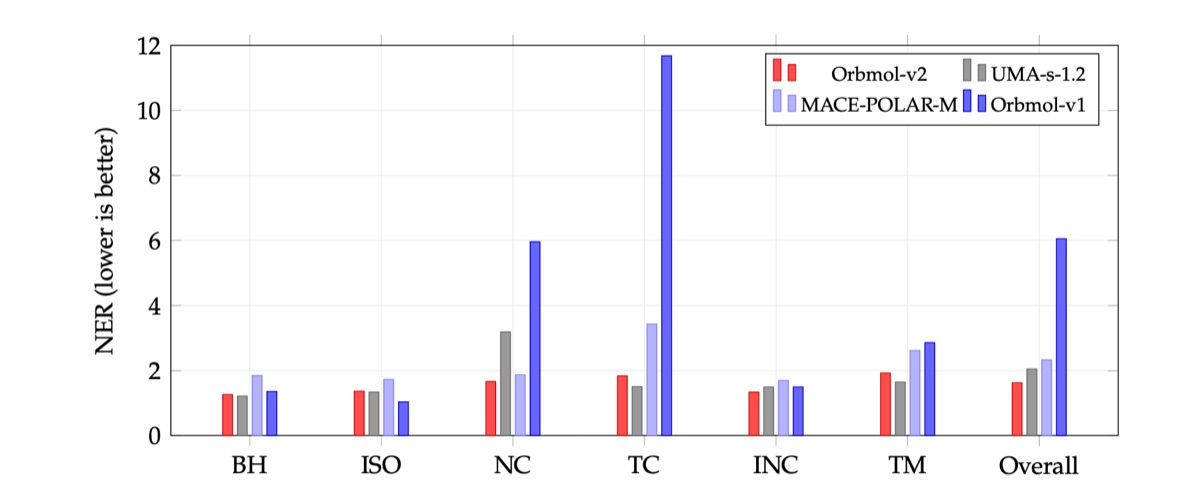
Figure 3: Per-category GSCDB137 NER comparison. OrbMol-v2 achieves the lowest overall NER (1.62) and leads on intramolecular noncovalent interactions (INC) and transition metal chemistry (TM).
Dataset curation
MLIPs cannot evaluate isolated single atoms or non-energy properties (dipoles, polarizabilities, vibrational frequencies) that some GSCDB subsets target. For an apples-to-apples evaluation, we exclude 28 of 139 GSCDB subsets: 6 require non-energy quantities (electric-field response, vibrational frequencies, dipoles, hindered rotors, polarizabilities), and 22 require isolated-atom reference energies (atomization energies, ionization potentials, bond-dissociation, rare-gas monomer subsets). We additionally drop all reactions in the remaining subsets whose stoichiometry involves any single-atom species (the same limitation, applied at reaction level), and the two orphan subsets (SC74, OEEFD) absent from the GSCDB metadata file, leaving 5,152 reactions across 109 subsets identical for every MLIP and every DFT functional.
Architecture
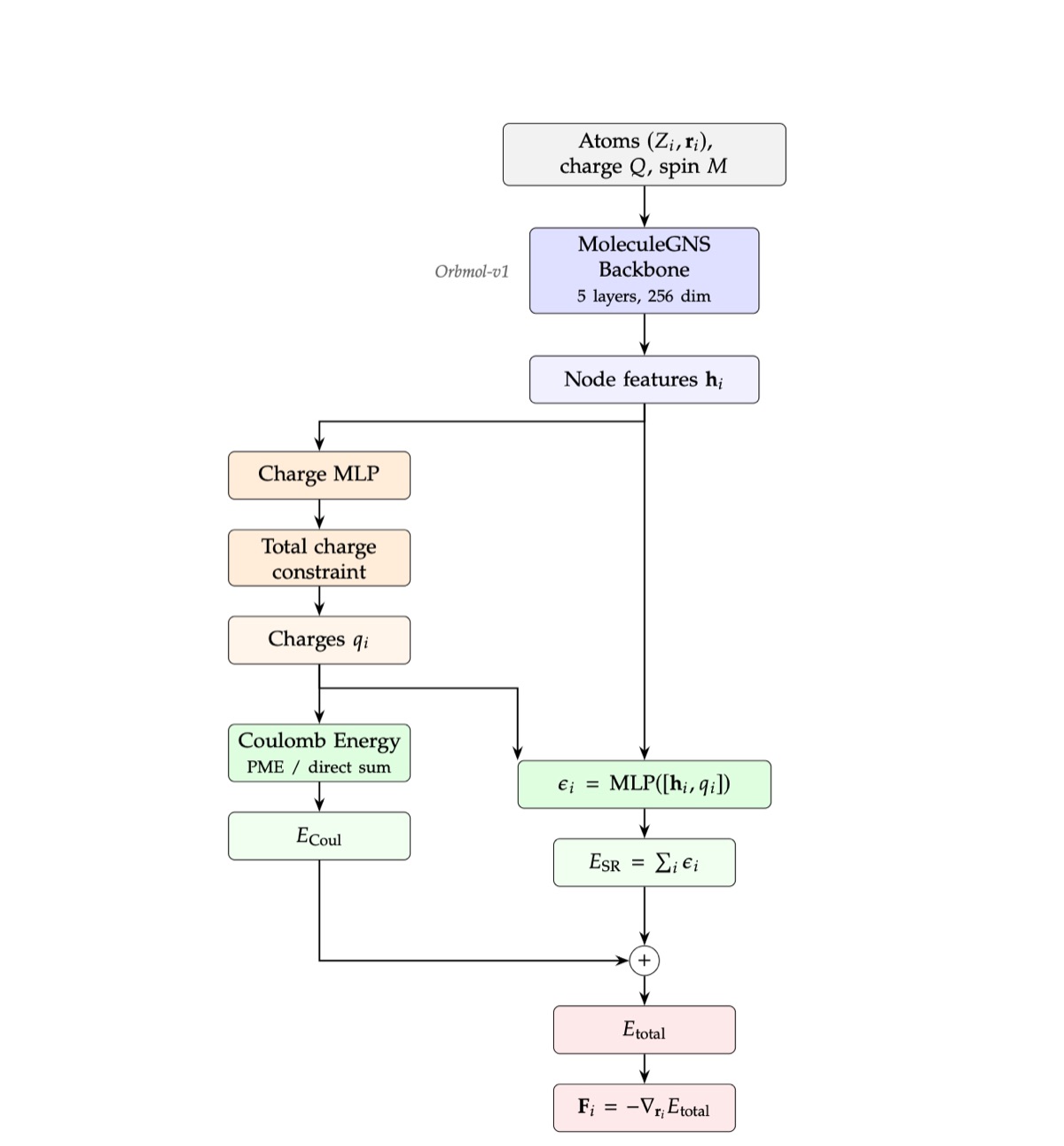
Figure 4: OrbMol-v2 architecture. The OrbMol-v1 backbone produces per-atom node features, which feed a charge prediction head. Predicted charges pass through a neutrality constraint enforcing Σᵢ qᵢ = Q. The charges feed into an explicit Coulomb energy (computed via PME for periodic systems or direct sum for molecules) and into a charge-conditioned energy head that also receives the backbone features. Charges are learned purely from energy and force supervision, with no explicit charge labels. Total spin multiplicity M enters the backbone as a global conditioning feature, as in OrbMol-v1. The total energy is the sum of the short-range and Coulomb terms, and forces are obtained by automatic differentiation.

